Chapter 2: Aromaticity and Resonance#
1. Introduction#
This section explores aromaticity, a fundamental concept in medicinal chemistry that profoundly influences a compound’s behavior. Aromaticity and resonance are two fundamental concepts that govern the stability, reactivity, and properties of organic molecules. In drug discovery, approximately 80% of approved drugs contain at least one aromatic ring. Understanding these concepts is essential for predicting molecular behavior, designing stable drug candidates, and interpreting structure-activity relationships.
2. Key Concepts and Definitions#
π (pi) Electrons: Electrons in p-orbitals that overlap sideways to form π bonds above and below the plane of the molecule.
Electron Delocalization: The spreading of electron density across multiple atoms rather than being confined between two atoms.
Resonance: Individual Lewis structures that differ only in electron placement (not atom positions). The actual molecule is a weighted average of all resonance contributors.
Aromaticity: Aromaticity: Exceptional stability arising from cyclic delocalization of π electrons following Hückel’s rule (4n+2 π electrons).
3. Main Content#
3.1 Understanding Resonance#
In 1865, Friedrich Kekulé proposed a structure for benzene (C₆H₆) with alternating single and double bonds. However, this structure created a puzzle:
Bond length uniformity: X-ray crystallography shows all six C-C bonds are 1.39 Å—exactly between a single bond (1.54 Å) and double bond (1.34 Å).
Unusual stability: If benzene had three isolated double bonds, we’d expect certain reactions. Instead, benzene is remarkably unreactive to addition reactions that alkenes readily undergo.
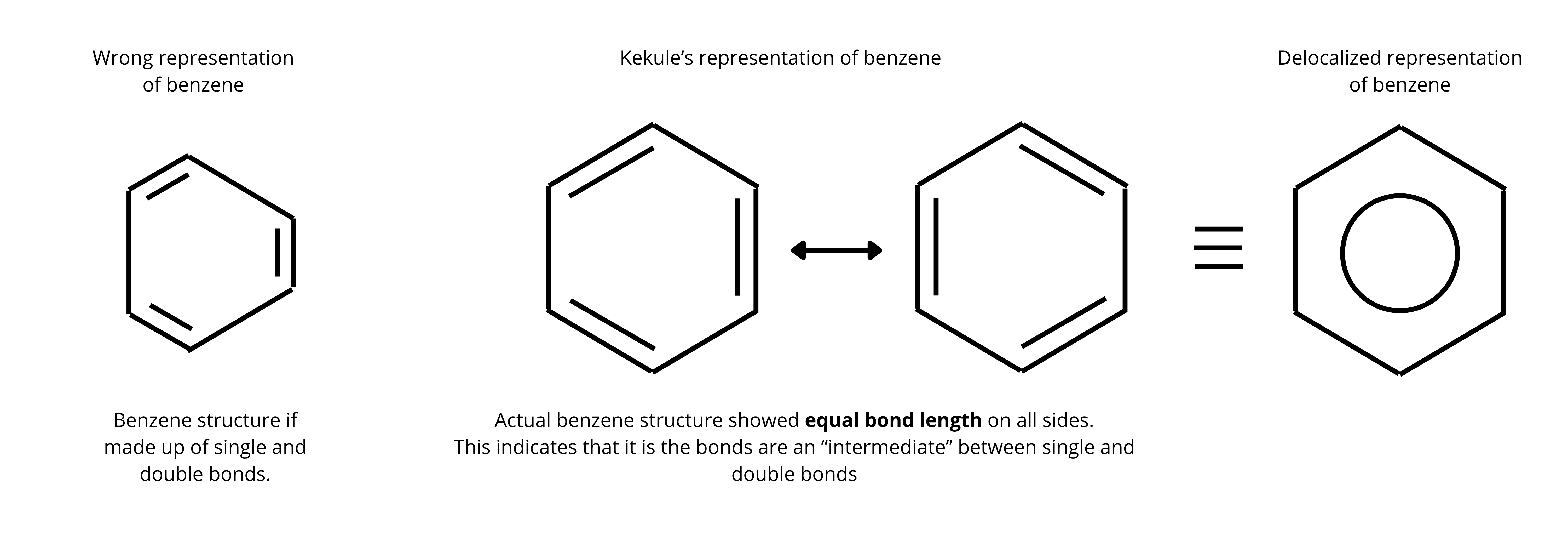
The answer lies in understanding how electrons are actually distributed in benzene.
In reality, each carbon atom contributes one electron in its p-orbital. With 6 carbons, that’s 6 electrons that can be delocalized in the ring.
The mechanism of delocalization:
p-Orbital overlap: The six p-orbitals (one from each carbon) are all parallel and can overlap with their neighbors.
Continuous π system: As the p-orbitals overlap around the entire ring, this creates a continuous electron cloud. We call this the π system
Electron cloud formation: The 6 π electrons occupy molecular orbitals that extend over all six carbons—above and below the ring plane like two donuts.
Energy lowering: This delocalization lowers the energy of the system, making benzene more stable than expected. This extra stability is called resonance energy or delocalization energy (~36 kcal/mol for benzene).
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
The phenomenon where electrons are delocalized is known as resonance.
3.2 Aromaticity#
Now that we understand benzene’s electron delocalization (resonance), we can extend to the concept of aromaticity.
Aromaticity is the special stability that arises when:
A cyclic system The molecule must form a closed ring to create a continuous loop for electron delocalization. Without a ring, electrons cannot circulate completely around the system, preventing the formation of the stabilizing aromatic electron cloud.
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
Contains a fully conjugated π system (continuous overlap) that exhibits resonance
Every atom in the ring must have a p-orbital that overlaps with its neighbors without interruption, creating an unbroken “highway” for π electrons to delocalize. If even one atom lacks a p-orbital (like sp³ hybridized carbons in cyclohexane), the conjugation is broken and aromaticity is lo
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
Has a planar geometry (to allow p-orbital overlap) All atoms in the ring must lie in the same plane so their p-orbitals can align parallel to each other and overlap effectively above and below the ring. If the ring is puckered or twisted, the p-orbitals point in different directions and cannot overlap properly, breaking the conjugation.
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
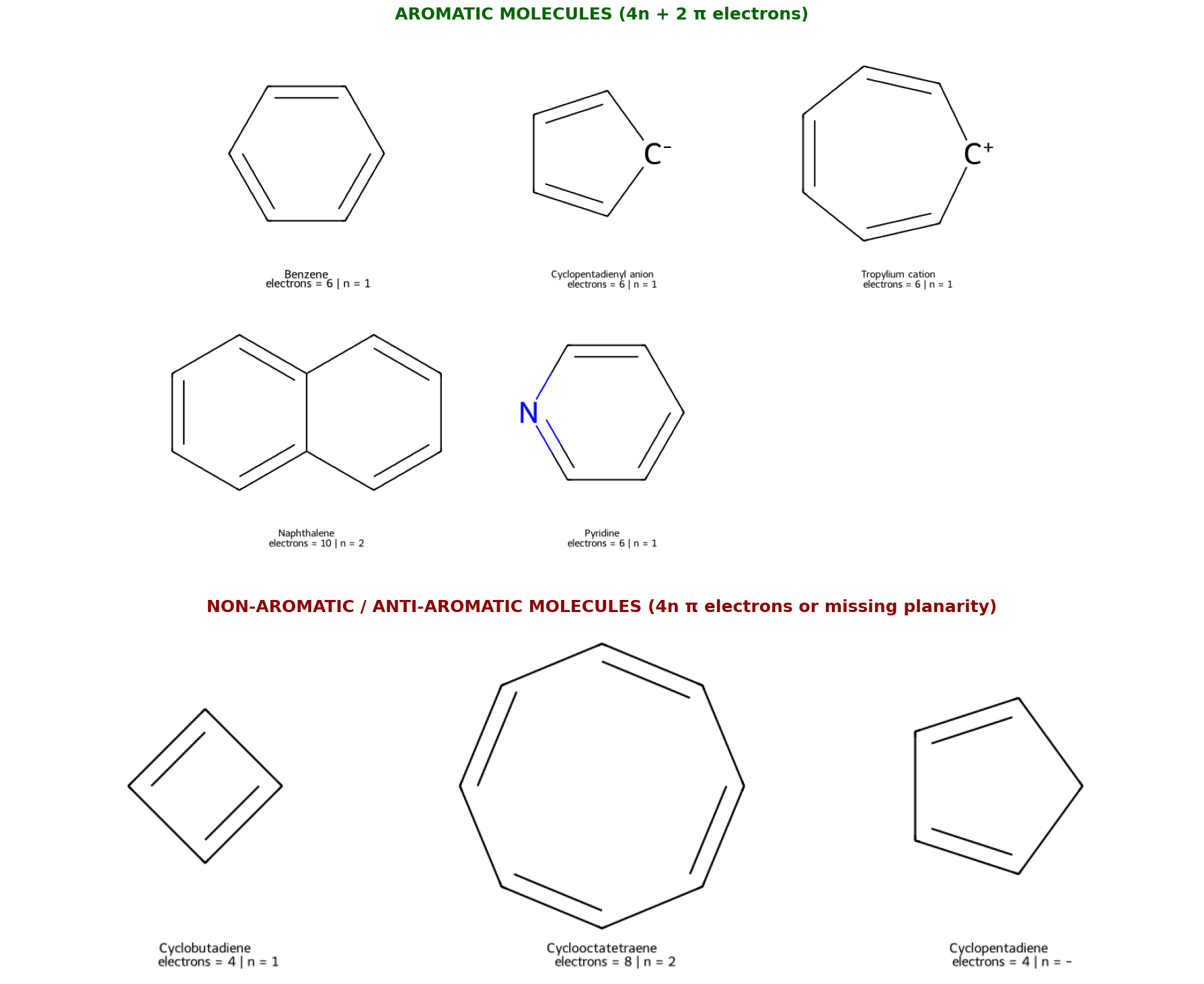
Follows Hückel’s rule: 4n+2 π electrons (where n = 0, 1, 2, 3…)
The system must contain a specific “magic number” of π electrons that completely fills all bonding molecular orbitals. This creates maximum stabilization.
Molecule |
π Electrons |
n Value |
4n or 4n+2? |
Classification |
|---|---|---|---|---|
Benzene |
6 |
1 |
4n + 2 |
Aromatic ✅ |
Cyclopentadienyl anion |
6 |
1 |
4n + 2 |
Aromatic ✅ |
Tropylium cation |
6 |
1 |
4n + 2 |
Aromatic ✅ |
Naphthalene |
10 |
2 |
4n + 2 |
Aromatic ✅ |
Pyridine |
6 |
1 |
4n + 2 |
Aromatic ✅ |
Cyclobutadiene |
4 |
1 |
4n |
Anti-aromatic ❌ |
Cyclooctatetraene |
8 |
2 |
4n |
Non-aromatic ❌ |
Cyclopentadiene |
4 |
- |
- |
Non-aromatic ❌ |
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
4. Summary and Key Takeaways#
In this section, we’ve explored the concept of aromaticity and its critical role in drug design and function. We learned to identify aromatic systems using Hückel’s rule.
Benzene’s six π electrons are not localized between carbon pairs but spread continuously across all six carbons through overlapping p-orbitals. This phenomenon is called resonance
Aromaticity is a special type of cyclic resonance requiring four criteria: (1) cyclic structure, (2) planar geometry, (3) fully conjugated π system, and (4) 4n+2 π electrons (Hückel’s rule). When all criteria are met, the molecule gains exceptional stability
Understanding aromaticity is essential for interpreting how a molecule binds to its target and how it will be processed by the body.