Chapter 2: Hybridization and Molecular Geometry#
1. Introduction#
Welcome to the study of hybridization and molecular geometry, where we translate flat, 2D chemical drawings into the 3D shapes that govern their biological activity. The central goal of this section is to learn how to predict an atom’s local 3D geometry from its hybridization. This skill is fundamental in medicinal chemistry, as the precise shape of a drug molecule determines whether it can successfully bind to its protein target, much like a key fitting into a lock. Our learning journey will begin with the rules for determining hybridization, then explore the specific geometries of sp³, sp², and sp hybridized atoms, and finally apply this knowledge to understand how the shapes of real drug molecules like Ritalin and Acetaminophen are critical to their function.
The animation below demonstrates that molecules are not planar. As the molecule adopts a stable 3D conformation, its energy drops. This spontaneous transition to a 3D shape proves that molecular bonds must be angled.
Cyclodecane | C10H20 | Target: 25.0 kcal/mol
2. Key Concepts and Definitions#
Hybridization: The mixing of atomic orbitals to form new hybrid orbitals with specific geometries, enabling atoms to form bonds with defined spatial arrangements.
Molecular Geometry: The three-dimensional arrangement of atoms in a molecule, determined by hybridization state and electron pair repulsion.
sp³ Hybridization: One s and three p orbitals mix to form four equivalent hybrid orbitals arranged tetrahedrally (~109.5° bond angles).
sp² Hybridization: One s and two p orbitals mix to form three equivalent hybrid orbitals arranged trigonally (~120° bond angles), with one unhybridized p orbital for π bonding.
sp Hybridization: One s and one p orbital mix to form two equivalent hybrid orbitals arranged linearly (180° bond angles), with two unhybridized p orbitals for π bonding.
Lone Pairs: Non-bonding electron pairs that occupy hybrid orbitals and affect molecular geometry.
3. Main Content#
3.1 Atomic Orbitals#
Every atoms have atomic orbitals. Formally, atomic orbitals are mathematical functions describing electron probability distributions, each with distinct shapes and energies that determine bonding capability.
In plain English, atomic orbitals are region of space that contain electrons. The common orbitals important in our work would be the s orbitals and p orbitals
s Orbitals
Spherically symmetric
Can hold 2 electrons
p Orbitals
Dumbbell-shaped with two lobes
Each can hold 2 electrons
Three types of p orbitals (px, py and pz)
Each of these orbitals are orthogonal (meaning 90°) to each other
d Orbitals
Complex shapes (four-lobed or doughnut)
Five types of d orbitals (dxy, dxz, dyz, dx²-y², dz²)
RARELY involved in organic medicinal chemistry
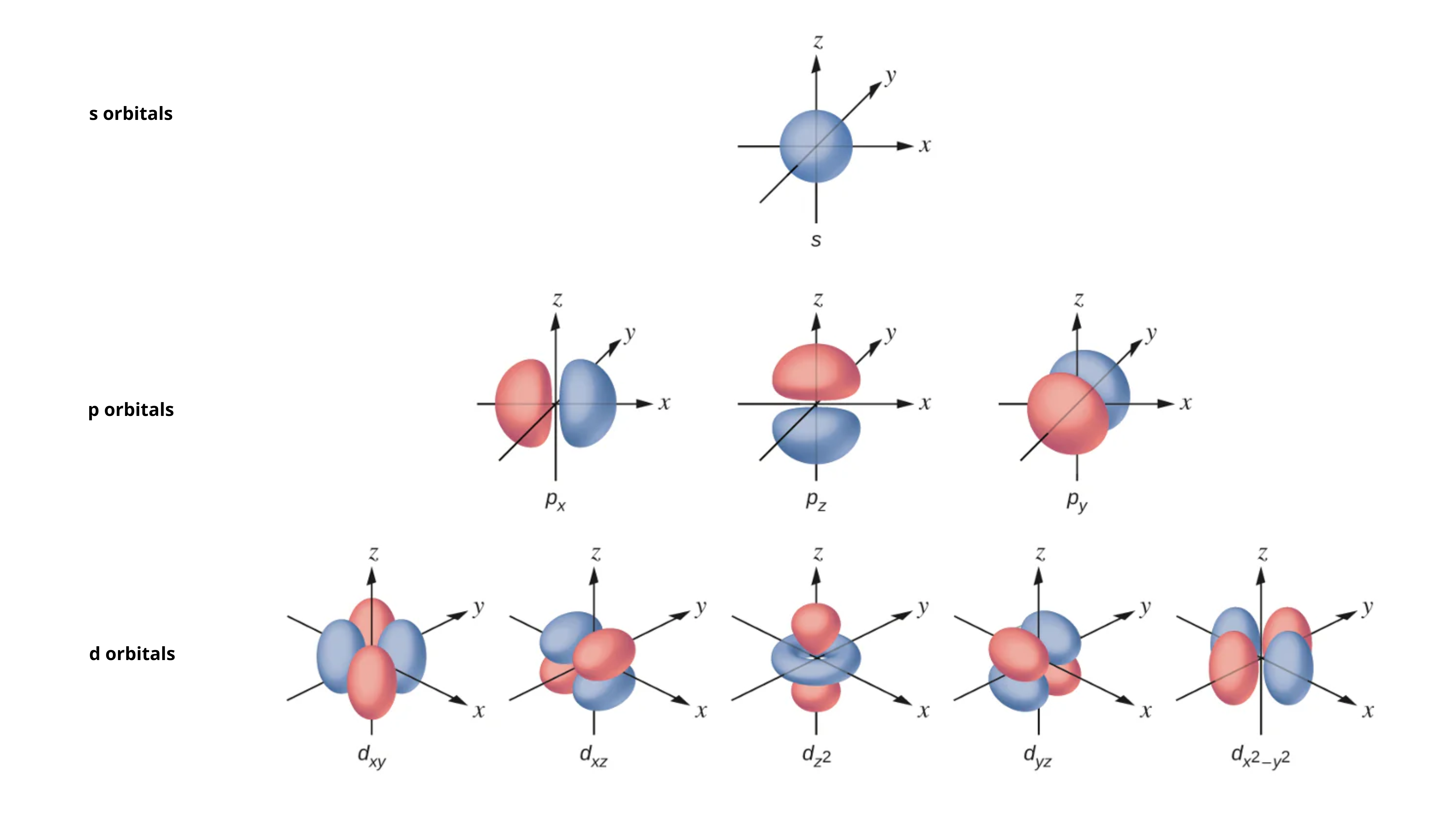 Figure adapted from source
Figure adapted from source
These atomic orbitals can be mixed to form new molecular orbitals with different shapes.
We call this mixing process hybrdization
For example, an s orbital (atomic orbital) can hybridize (mix) with a p orbital (atomic orbital) to form a NEW sp orbital (molecular orbital)
3.2 Hybridization and Molecular Orbitals#
The ability of atoms like carbon, oxygen and nitrogen to adopt different hybridization states creates structural diversity in drug molecules. Each hybridization state produces distinct geometries that affect how molecules fit into binding sites.
3.2.1 sp³ Hybridization (Tetrahedral)#
An atom with four electron domains adopts sp³ hybridization, leading to a tetrahedral arrangement.
Electron Domains: 4
Electron Geometry: Tetrahedral
Molecular Geometry: Tetrahedral (with 0 lone pairs)
Ideal Bond Angle: ~109.5°
3.2.2 sp² Hybridization (Trigonal Planar)#
An atom with three electron domains is sp² hybridized, resulting in a flat, trigonal planar geometry.
Electron Domains: 3
Electron Geometry: Trigonal Planar
Molecular Geometry: Trigonal Planar (with 0 lone pairs)
Ideal Bond Angle: ~120°
3.2.3 sp Hybridization (Linear)#
An atom with two electron domains is sp hybridized, yielding a linear geometry.
Electron Domains: 2
Electron Geometry: Linear
Molecular Geometry: Linear (with 0 lone pairs)
Ideal Bond Angle: 180°
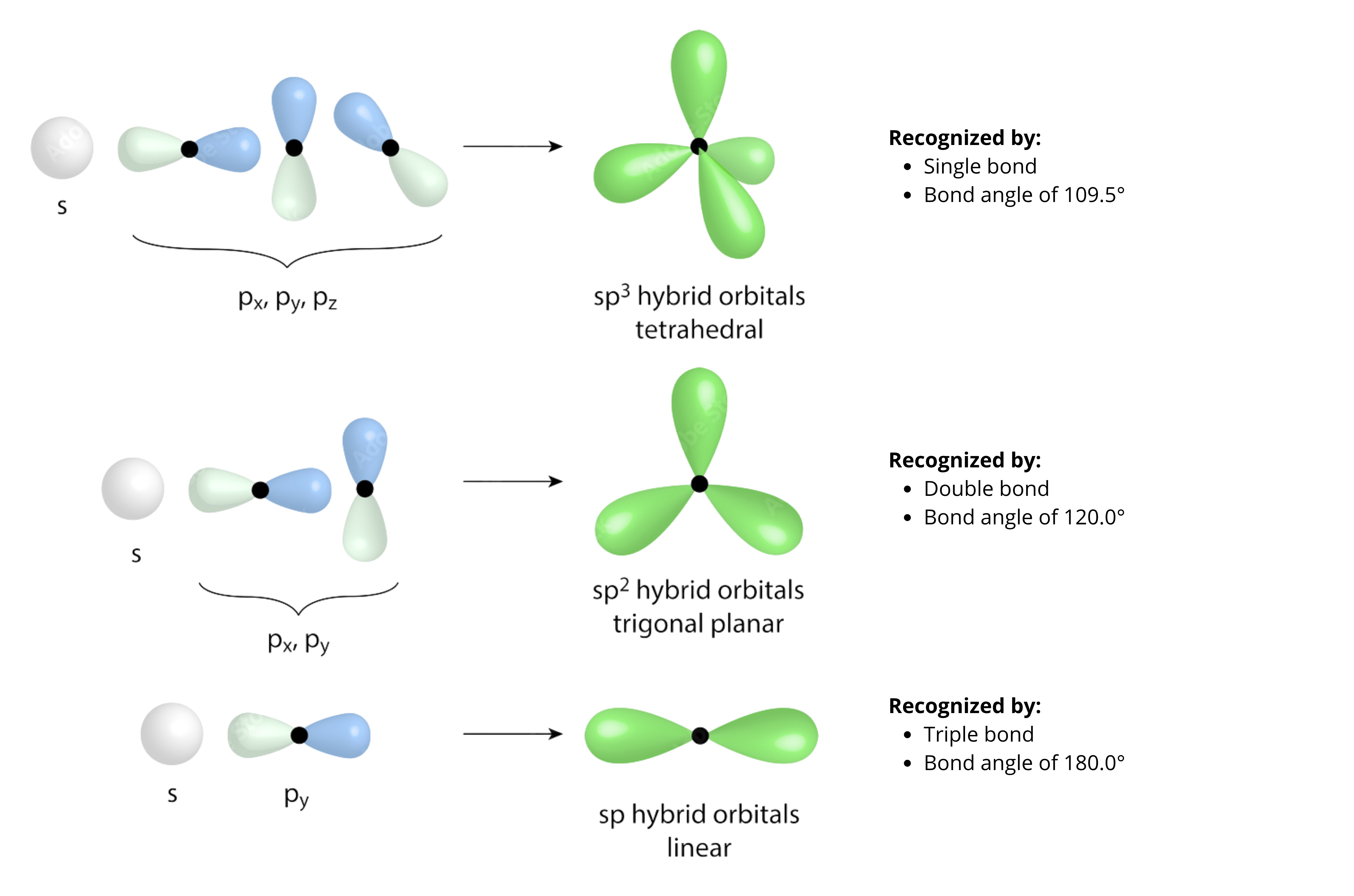 Figure adapted from source
Figure adapted from source
By looking at the type of bonds, you would be able to tease out what is the hybridization state of the atom, and from there deduce the likely shape based on the bond angles.
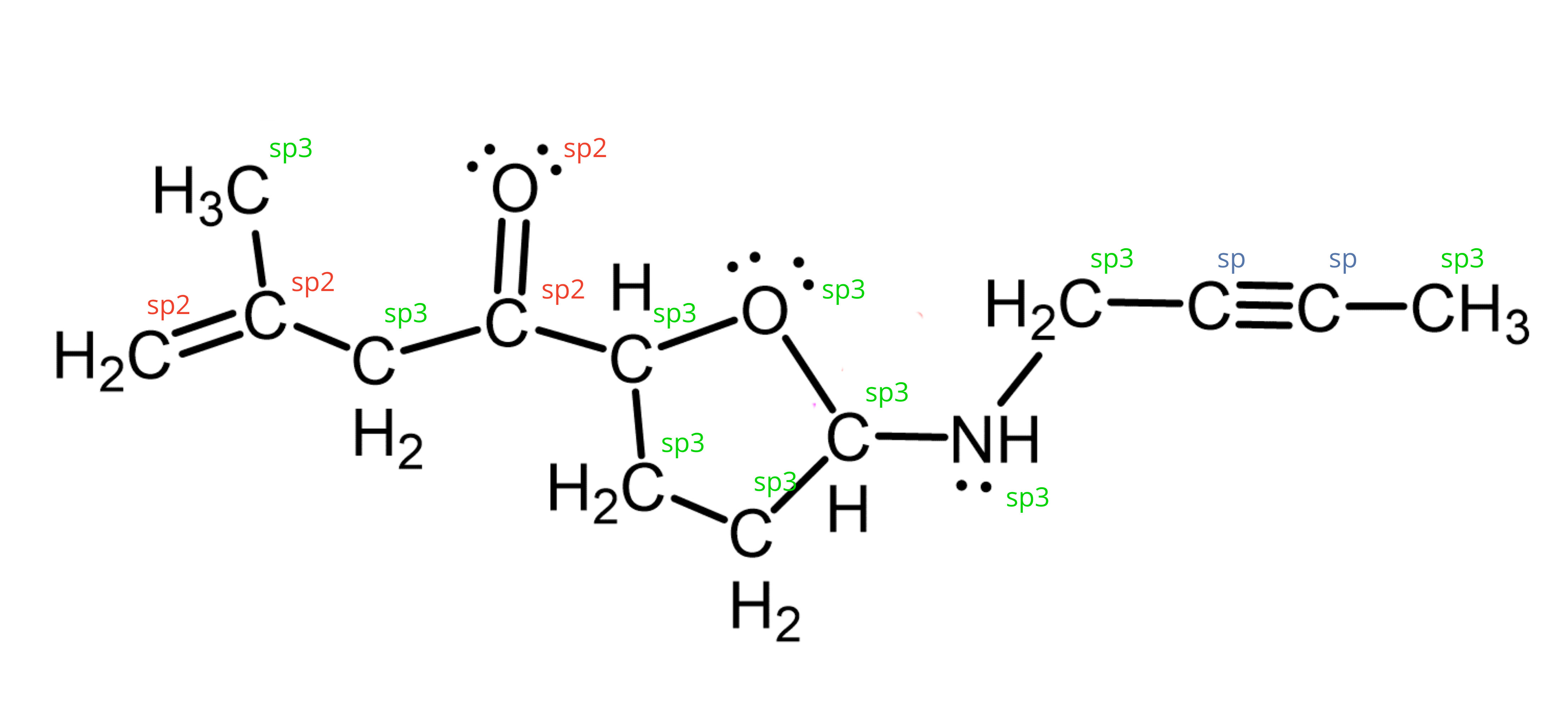 Figure adapted from source
Figure adapted from source
Distortion of Bond Angles like Lone Pair#
3.4 The Impact of Lone Pairs on Heteroatoms#
The presence of lone pairs on heteroatoms (like N and O) causes the molecular geometry (atom arrangement) to differ from the electron geometry (domain arrangement).
Hybridization |
Electron Domains |
Lone Pairs |
Electron Geometry |
Molecular Geometry |
Approx. Bond Angle |
|---|---|---|---|---|---|
sp³ |
4 |
0 |
Tetrahedral |
Tetrahedral |
~109.5° |
sp³ |
4 |
1 |
Tetrahedral |
Trigonal Pyramidal |
~107° |
sp³ |
4 |
2 |
Tetrahedral |
Bent |
~104.5° |
Important: Lone pairs are more repulsive than bonding pairs. This electron-electron repulsion “pushes” the bonding pairs closer together, compressing the bond angles from the ideal (e.g., 109.5° in a perfect tetrahedron) to smaller values (~107° in ammonia, ~104.5° in water). This distortion directly impacts how a molecule fits into a binding site.
sp³ Nitrogen: An amine nitrogen has four electron domains (3 bonds, 1 lone pair). The lone pair’s repulsion compresses bond angles to ~107°, creating a trigonal pyramidal shape. This positions the lone pair to act as a potent hydrogen bond acceptor, as seen in Ritalin.
sp³ Oxygen: An alcohol oxygen has four electron domains (2 bonds, 2 lone pairs). The two lone pairs compress the bond angle to ~104.5°, resulting in a bent geometry, which precisely directs its lone pairs for binding, as in Acetaminophen’s hydroxyl group.
sp² Oxygen: A carbonyl oxygen has three electron domains (1 double bond, 2 lone pairs), which adopt a trigonal planar electron geometry with ~120° angles, consistent with sp² hybridization. This geometry is critical for directing hydrogen bonds, as seen in Acetaminophen’s carbonyl.
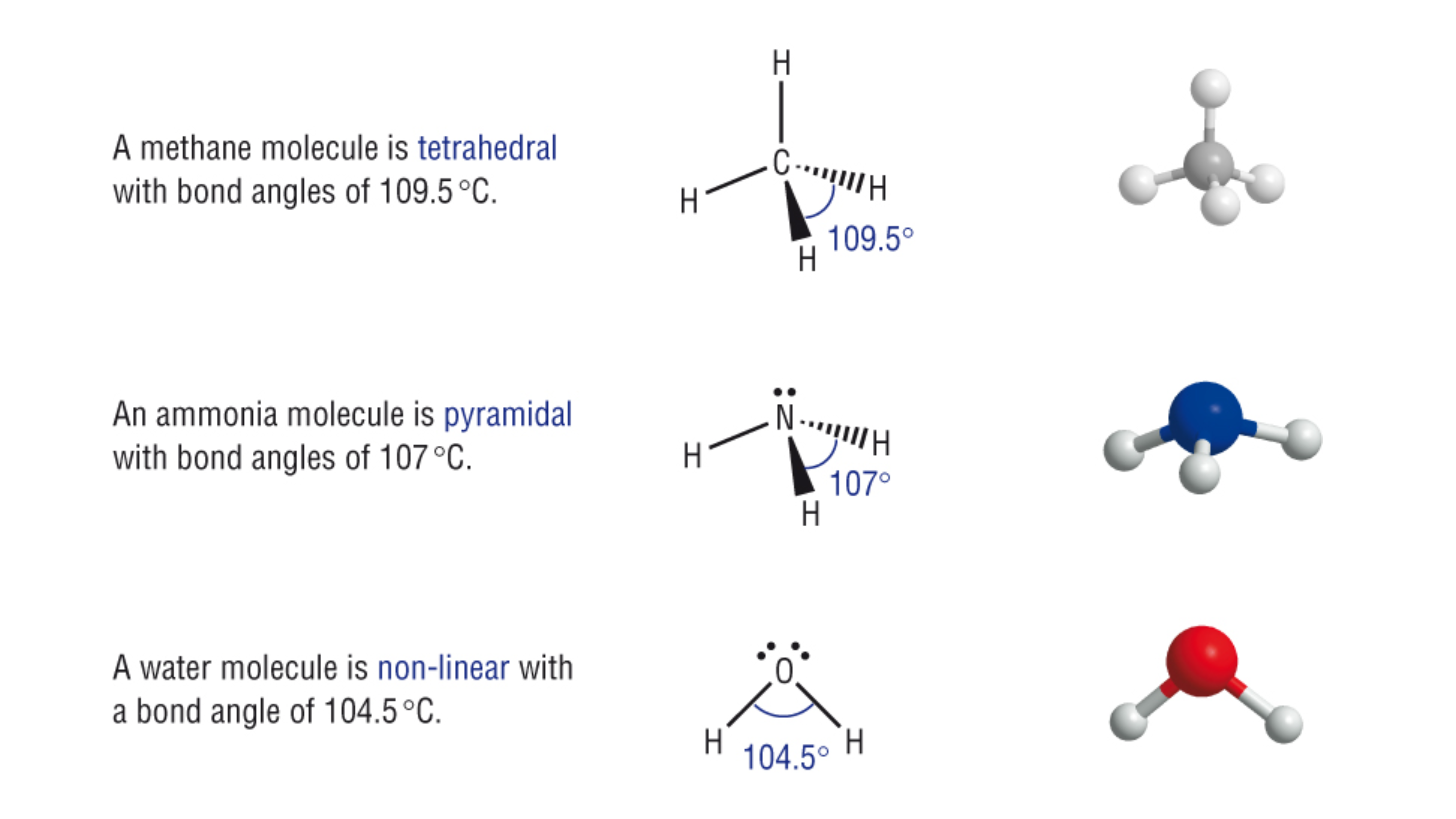 Figure adapted from source
Figure adapted from source
3.5 Special Case: The Planar Amide Nitrogen#
Unlike a typical sp³ pyramidal amine, an amide nitrogen is a key exception. Due to resonance with the adjacent carbonyl, its lone pair delocalizes, forcing a flat, sp² trigonal planar geometry.
Important: The planarity of the amide bond is a fundamental principle in protein structure. This rigidity, caused by resonance, prevents free rotation and forms the backbone of peptides and proteins, dictating how they fold into complex 3D structures like alpha-helices and beta-sheets. Understanding this exception is crucial for understanding drug-protein interactions.
Notice in the animation below that N-methylacetamide with an amide group (-NHCO) is planar, while dimethylamine with an amine group (-NH) group is not.
3Dmol.js failed to load for some reason. Please check your browser console for error messages.
4. Summary and Key Takeaways#
In this section, we’ve explored how the concept of orbital hybridization allows us to predict the three-dimensional geometry of molecules, a critical skill for understanding drug-target interactions. We have seen that the shape of a molecule is rarely accidental; it is a direct consequence of the electronic structure of its atoms.
The number of electron domains (bonds and lone pairs) around an atom determines its hybridization: 4 domains = sp³, 3 domains = sp², and 2 domains = sp.
Hybridization dictates the electron geometry (tetrahedral, trigonal planar, linear), while the presence of lone pairs determines the final molecular geometry (e.g., trigonal pyramidal, bent).
Lone pairs are sterically demanding, compressing bond angles and serving as crucial hydrogen bond acceptors in biological systems.
Special cases like amides demonstrate that resonance can enforce planarity, overriding simple hybridization rules and creating rigid structural motifs essential for drug activity.